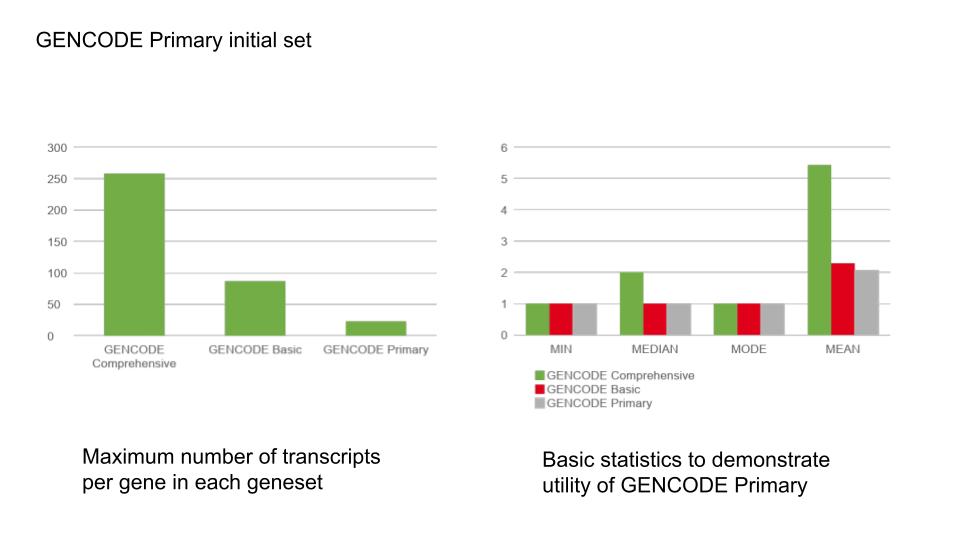
The GENCODE Primary transcript set
Long-read sequencing technologies such as those developed by Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) have driven a step-change in our ability to capture the transcriptome. As a reference gene annotation resource GENCODE aims to capture this transcript diversity in human and mouse and present it in an organised way to support its use in downstream analysis.
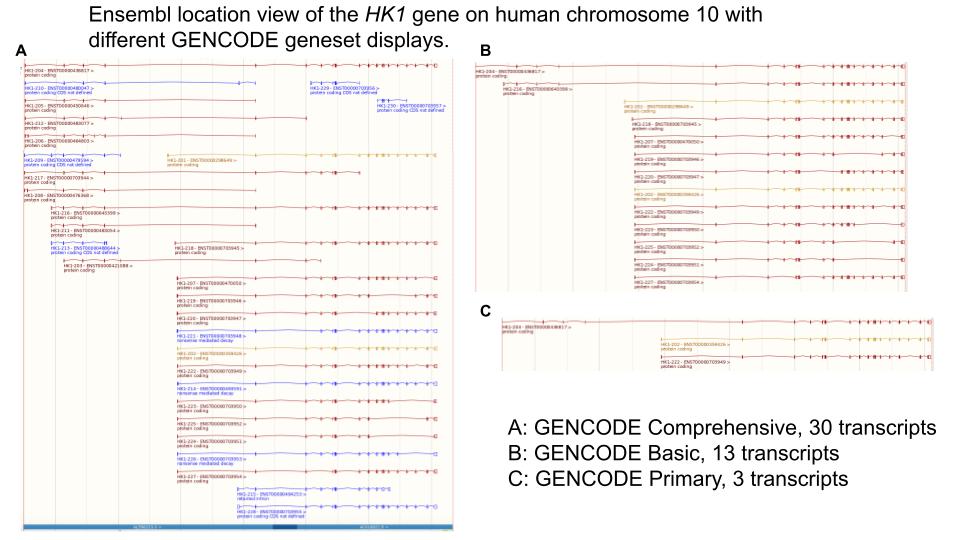
Historically, we have presented two categories of transcripts in GENCODE gene annotation: GENCODE Comprehensive, which captures all annotated transcripts, and GENCODE Basic, a smaller subset of transcripts containing only full-length transcripts at protein-coding genes and enriched for alternative splicing at lncRNA genes. The length and depth of the reads generated in ONT and PacBio experiments will eventually overwhelm the GENCODE Basic category, for example by adding many tens or hundreds of full-length alternatively spliced transcripts at protein-coding genes. While this rich set of annotations will benefit many use-cases, we recognise that very many transcripts can hinder others, not least viewing GENCODE transcripts alongside other data in the Ensembl and UCSC genome browsers. As such we will be changing the way transcripts are represented, adding a new classification with a small subset of GENCODE Comprehensive transcripts enriched for signals of functional potential.
The GENCODE Primary transcripts capture a minimal set of transcripts at protein coding genes and include protein-coding exons with evidence of evolutionary constraint and alternative splicing (including exon skips) with high expression. The GENCODE Primary subset includes all MANE Select/Ensembl Canonical and MANE Plus Clinical transcripts by default.
For the human reference genome, in Ensembl 113/GENCODE 47, GENCODE Primary flags will be available on the Ensembl website and in release files, while the default annotation displayed in the Ensembl browser will be GENCODE Basic. In Ensembl 114/GENCODE 48 the default annotation displayed in the Ensembl browser will be GENCODE Primary. The annotation sets GENCODE Comprehensive and GENCODE Basic can be added to the browser.
The GENCODE Primary set provides an opportunity to simplify variant interpretation by reducing the number of variant transcript predicted molecular consequences needing review. These data are only now displayed on variant and transcript pages in the browser for GENCODE Primary transcripts. Annotation of your own variants with the Ensembl Variant Effect Predictor now also takes advantage of this transcript set. Annotation can be restricted to only these transcripts, all transcripts (the GENCODE Comprehensive set), or GENCODE Basic transcripts.
Mouse has no MANE Select transcripts annotated. However, we are developing a GENCODE Primary pipeline for mouse based on the Ensembl Canonical transcript set and we anticipate adding GENCODE Primary annotation in Ensembl 115/GENCODE M38.
Unlike MANE transcripts, which are very stable, the GENCODE Primary set is expected to be dynamic over the initial releases due to both the addition of new transcript models based on long transcriptomic data and active pipeline development to capture all relevant transcripts at protein-coding genes and lncRNAs.
GENCODE Primary transcripts at protein-coding genes are made with reference to the MANE Select transcripts [1]. MANE Plus Clinical transcripts are added to the GENCODE Primary set by default.
Starting with the GENCODE Comprehensive annotation:
- MANE Select and MANE Plus Clinical transcripts are added to GENCODE Primary set
- all incomplete transcripts are removed from consideration
- all transcripts with no annotated CDS (biotypes: retained_intron, protein_coding_CDS_not_defined) are removed from consideration
- each remaining transcript is compared in turn to the MANE Select transcript
- all intron and exon features not present in MANE Select are identified
- terminal exon subsequences are considered as present in MANE Select
- novel exons are evaluated for evolutionary constraint via simple overlap with PhyloCSF candidate coding regions [2, 3]
- constrained exons are retained
- novel (non-MANE) introns are evaluated for inclusion using Recount3 [4]
- the ratio between the Recount3 read count of the novel intron and the mean read count of all overlapping MANE introns is calculated
- analogous to Junction Inclusion Ratio (JIR) calculations [5]
- novel introns with inclusion ratio >1:20 (>5%) are retained
- retained novel exons and introns are collapsed to remove redundancy
- transcripts containing retained novel exons and introns are identified
- transcripts are ranked using a pipeline developed to identify Ensembl/GENCODE MANE Select candidate transcripts [1]
- the highest ranking transcript containing a retained novel exon/intron is added to the GENCODE Primary set
- this process is iterated until all retained novel exons and introns are represented
All GENCODE Primary transcripts will be manually reviewed to validate their selection (this work is currently in progress).
For all biotypes other than protein-coding genes in human, the GENCODE Primary flag is added to the Ensembl Canonical transcript in Ensembl 114/GENCODE 48, and for lncRNA genes only this will be the transcripts with the longest genomic span. We are working on a specific lncRNA pipeline to enable the addition of relevant highly expressed transcripts to the set.
References
- Morales J, Pujar S, Loveland JE, et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature. 2022 Apr;604(7905):310-315. DOI: 10.1038/s41586-022-04558-8. PMID: 35388217; PMCID: PMC9007741.
- Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics (Oxford, England). 2011 Jul;27(13):i275-82. DOI: 10.1093/bioinformatics/btr209. PMID: 21685081; PMCID: PMC3117341.
- https://data.broadinstitute.org/compbio1/PhyloCSFtracks/trackHub/hub.DOC.html
- Wilks C, Zheng SC, Chen FY, et al. recount3: summaries and queries for large-scale RNA-seq expression and splicing. Genome Biology. 2021 Nov;22(1):323. DOI: 10.1186/s13059-021-02533-6. PMID: 34844637; PMCID: PMC8628444.
- Nellore A, Jaffe AE, Fortin JP, et al. Human splicing diversity and the extent of unannotated splice junctions across human RNA-seq samples on the Sequence Read Archive. Genome Biology. 2016 Dec;17(1):266. DOI: 10.1186/s13059-016-1118-6. PMID: 28038678; PMCID: PMC5203714.